الثلاسيميا هو أحد امراض الدم الوراثية – يوجد في العالم اكثر من 100 مولود جديد يولدون مصابين بهذا المرض بأنوعه المختلفة بدرجاته المختلفة – كل عام
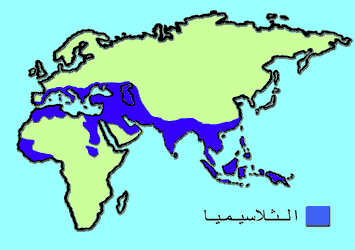
تنتشر الثلاسيميا بشكل اكثر في سكان ايطاليا – اليونان – دول حوض البحر المتوسط – شمال قارة آسيا – والدول الافريقية المحاذية للبحر المتوسط
———————————–
ما هي انواع الثلاسيميا المختلفة :-
تتشكل الثلاسيميا في انواع مختلفة من فقر الدم ( فشل كريات الدم الحمراء في العمل بشكل طبيعي )
بحسب المجموعتين الرئيسيتين من مجموعات الدم
تسمى
1- مجموعة ثلاسيميا ألفا
و 2- مجموعة ثلاسيميا بيتا
وتعتمد التسمية على تحديد اي نوع نوع من حاملات الاكسجين ( الهيموجلوبين ) يوجد خلل في عمله
أكثر ألأنواع شدة وتأثيراً تلك المنتشرة في دول جنوب شرق آسيا مثل الصين – والفلبين وتسمى ثلاسيميا الفا وتتسبب في ولادة طفل ميت – ان معظم ألأشخاص المصابين بهذا المرض يعانون من فقر الدم متنوع الدرجات
—————————–
نركز هنا في هذه النشرة على النوع المعروف بثلاسيميا بيتا والتي تختلف درجاتها في الشدة من شديدة جدا الى بدون أثر على الصحة
1- الثلاسيميا الكبرى
اكثر انواع الثلاسيميا بيتا شدة وتأثيرا على المريض والتي تسمى ايضا ب كولي انيميا ( فقر الدم الذي وصفه الدكتور كولي عام 1925
2- الثلاسيميا المتوسطة وهي ايضا من النوع المتوسط الذي وصفه الدكتور كولي
3- الثلاسيميا الصغرى والتي تسمى ثلاسيميا تريت وقد لا تتسبب في ظهور اية اعراض على المريض ولكن التغيرات في الدم قابلة للحصول
—————————————————-
كيف تؤثر ألاصابة بالمرض على الطفل :-
ان معظم ألأطفال المصابيت بالثلاسيميا الكبرى يظهرون كأنهم أصحاء تماماً عند الولادة ولكن وخلال السنة الاولى من الولادة او السنتين ألأوليين يبدأ لون البشرة في الميل الى الشحوب ثم تبدأ أعراض فقدان الشهية للطعام والبطء في النمو وتأخذ البشرة في الاصفرار واذا بقي الطفل المريض بدون علاج فان الطحال والكبد يتضخمان ( يزداد حجمهما ) ثم تبدأ العظام في الهشاشة والتقوس وخاصة عظام الوجه وأطفال الثلاسيميا غالبا ما يشبهون بعضهم البعض في المظهر
ان فشل وظائف القلب وتكرر الاصابة بالالتهابات هي أحد أكثر اسباب الوفاة عند هؤلاء ألأطفال شيوعا
———————————
ألأطفال المصابون بالثلاسيميا الوسطى قد يعانون من نفس اعراض الثلاسيميا الكبرى على الرغم من انه وفي معظم الحالات فان آثار المرض تكون خفيفة في السنوات العشرون الاولى من الاصابة بالمرض
———————————–
كيف ينتقل المرض :-
ان الثلاسيميا بكل انواعها مرض وراثي اي انها تنتقل بواسطة الوراثة فقط ولا يمكن الاصابة بها بالعدوى من طفل الى آخر فالمرض ينتقل عبر ألأبوين الحاملين للجين المصاب فقط الذين يورثانه لأولادهما فحامل المرض يحمل جين وراثي سليم وغير مصاب بالمرض وجين آخر مصاب به وهو الوضع الذي يسمى احيانا تلسيميا تريتي ومعظم حاملي المرض يعيشون حياة طبيعية ويتمتعون بصحة جيدة
عندما يتزوج شخصين حاملين للمرض وينجبون ألأبناء فان طفل واحد من بين كل اربعة مواليد لهم يولد حامل لجين مصاب بالثلاسيميا فيكون حاملا للمرض ومولود واحد من كل اربع مواليد يحمل الجينين السليمين فيأتي صحيحا معافى من المرض تماما والمولودين الآخرين يوادون اما حاملا للمرض او مصابا به بالكامل
———————————
هل يوجد فحص يمكن ان يحدد :-
نعم يوجد – ففحص الدم للأسرة واجراء الدراسات الوراثية على افراد ألأسرة يمكن ان تظهر فيما اذا كان فرد او افراد من الاسرة مصابين او حاملين للجين الوراثي الخاص بالمرض
ان التشخيص المبكر والسابق للزواج يعتبر امر حيوي ومهم جدا لتجنب انجاب أطفال مصابين بهذا المرض العضال
—————————–
ما العلاج :-
يتمثل علاج ألأشخاص المصابين بالثلاسيميا على مرحلتين
1- العلاج الفوري والذي يسمى علاجا تجاوزا للحقيقة فهو مجرد تأجيل للأجل المحتوم ويتمثل في
نقل الدم المتكرر وتعاطي انواع مختلفة من المضادات الحيوية وخاصة بالنسبة لمرضى الثلاسيميا الكبرى
اما مرضى الثلاسيميا المتوسطة فهم عادة لا يحتاجون الى نقل الدم بصفة متكررة على الرغم من ان نقل الدم لهم قد يصبح حاجة ملحة اذا بدأت تعقيدات المرض بالظهور او اذا اوصى الأطباء بذلك
عندما يعالج اطفال الثلاسيميا الكبرى بنقل الدم المتكرر ( عادة كل 3 الى 4 اسابيع ) بهدف الاحتفاظ بمستوى عال للهيموجلوبين الناقل في اجسامهم فان الكثير من مضاعفات المرض يمكن تجنبها فان نمو الطفل يصبح طبيعيا ويمكن تجنب الفشل في أداء وظائف القلب واعوجاج او تقوس العظام
لسوء الحظ فان نقل الدم المتكرر يتسبب في ارتفاع نسبة الحديد المختزن في الجسم والذي قد ينتج عنه توقف القلب عن العمل او الكبد او اي عضو آخر من أعضاء الجسد
ان هؤلاء المرضى يجب ان يتعاطو نوع من ألأدوية التي تسمى طاردات الحديد يمكن ان تساعد المريض على التخلص من الحديد الزائد في جسمه مما يمنع أو يؤخر الاصابة بمضاعفات ارتفاع الحديد في الجسم
هذا النوع من الدواء يحقن عادة في جسد المريض بشكل يومي تحت الجلد بوساطة مضخة ميكانيكية أثناء نوم الطفل – ألطفال المصابين بالثلاسيميا الكبرى والذين يتلقون هذا العلاج – نقل الدم المتكرر وطاردات الحديد والمضادلت الحيوية قد يعيشون عشرون او ثلاثون عاما او اكثر
2- العلاج الدائم
يتمثل هذا العلاج في اجراء عملية زراعة للنخاع العظمي المسؤول عن تكوين الدم حيث يتخلص المريض من الجين الوراثي المسؤول عن المرض بصفة نهائية ولكن هذا النوع من العلاج متوفر فقط لاعداد قليلة من المرضى بسبب ارتفاع الكلفة المالية للعلاج واشتراط ( سابقا ) وجود متبرع مطابق للمريض
مع تمنياتي للجميع بدوام الصحة و العافية

.gif)
.gif)
.gif)
.gif)
.gif)
.gif)
.gif)
.gif)


